Objawy neurologiczne choroby Fanconiego u dzieci. Zespół Fanconiego: objawy i leczenie. Leczenie lekami
Zespół de Toni-Debre-Fanconi jest ciężki choroba wrodzona, charakteryzujący się różnorodnością dzieci cierpią na nią najczęściej w pierwszym roku życia. Zwykle spotykany w połączeniu z innymi patologie dziedziczne, ale może również objawiać się jako niezależny zespół.
Krótka wycieczka do historii
Chorobę odkrył i zbadał w 1931 roku dr Fanconi ze Szwajcarii. Badając dziecko z krzywicą, niskim wzrostem i zmianami w badaniach moczu, doszedł do wniosku, że tę kombinację objawów należy traktować jako oddzielną patologię. Dwa lata później de Tony dokonał własnych poprawek, dodał hipofosfatemię do już istniejącego opisu, a po pewnym czasie Debre ujawnił u podobnych pacjentów aminoacydurię.
W literaturze krajowej ten stan nazywa się terminami „ zespół dziedziczny de Toni – Debre – Fanconi” i „cukrzyca glukoaminofosforanowa”. Za granicą często określa się go mianem nerkowego zespołu Fanconiego.
Przyczyny zespołu Fanconiego
W ten moment Nie udało się w pełni dowiedzieć się, co leży u podstaw tej poważnej choroby. Zakłada się, że zespół Fanconiego to Specjaliści uważają, że rozwój tej patologii jest związany z mutacją punktową, która prowadzi do zła praca nerki. Liczne badania potwierdziły, że w organizmie dochodzi do naruszenia metabolizmu komórkowego. Możliwe, że w sprawę zaangażowany jest trójfosforan adenozyny (ATP) – związek, który odgrywa rolę ważna rola w wymianie energii. W wyniku nieprawidłowego funkcjonowania enzymów ginie glukoza, aminokwasy, fosforany i inne równie przydatne substancje. W tak trudnych warunkach kanaliki nerkowe nie otrzymują energii potrzebnej do funkcjonowania. Przydatny materiał wydalany z moczem, procesy metaboliczne są zaburzone, rozwijają się zmiany przypominające krzywicę tkanka kostna.
Zespół Fanconiego występuje znacznie częściej u dzieci niż u dorosłych. Według statystyk częstotliwość patologii wynosi 1: 350 000 noworodków. Zarówno chłopcy, jak i dziewczęta są dotknięci w równych proporcjach.
Objawy zespołu Fanconiego
Choroba może rozwijać się w każdym wieku, ale najczęściej występuje u dzieci w pierwszym roku życia. Glukozuria, uogólniona hiperaminoacyduria i hiperfosfaturia - ta triada znaków charakteryzuje zespół Fanconiego. Objawy rozwijają się dość wcześnie. Przede wszystkim rodzice zauważają, że ich dziecko zaczyna częściej oddawać mocz i jest ciągle spragnione. Niemowlęta oczywiście nie mogą tego powiedzieć słowami, ale przez ich kapryśne zachowanie i ciągłe wiszenie na klatce piersiowej lub butelce staje się oczywiste, że coś jest nie tak z dzieckiem.

W przyszłości rodzice przynoszą wiele niepokoju częstym bezprzyczynowym wymiotom, przedłużającym się i niewytłumaczalnym zaparciom.Z reguły na tym etapie dziecko w końcu trafia do lekarza. Doświadczony pediatra może podejrzewać, że ta kombinacja objawów wcale nie jest podobna do przeziębienia. Jeśli lekarz jest piśmienny, zdąży rozpoznać zespół Fanconiego na czas.
Tymczasem objawy nigdy nie ustępują. Dodaje się do nich zauważalne opóźnienie w rozwoju fizycznym i umysłowym, pojawia się wyraźna krzywizna dużych kości. Zwykle zmiany dotyczą tylko kończyn dolnych, prowadząc do deformacji typu szpotawości lub koślawości. W pierwszym przypadku nogi dziecka będą wygięte przez koło, w drugim - w kształcie litery „X”. Obie opcje są oczywiście niekorzystne dla przyszłego życia dziecka.
Zespół Fanconiego u dzieci często obejmuje osteoporozę (przedwczesna utrata masy kostnej) oraz znaczne opóźnienie wzrostu. Długie złamania i paraliż nie są wykluczone. Nawet jeśli do tej pory rodzice nie martwili się o stan dziecka, to na tym etapie na pewno nie odmówią wykwalifikowanej pomocy.

Zespół Fanconiego u dorosłych jest dość rzadki. Chodzi o to, że to poważna choroba naturalnie prowadzi do rozwoju niewydolność nerek. W tym scenariuszu nie można podać jednoznacznej prognozy i zagwarantować dużej.W piśmiennictwie opisano przypadki, gdy w wieku 7-8 lat zespół Fanconiego słabł, nastąpiła wyraźna poprawa stanu dziecka, a nawet powrót do zdrowia. Niestety takie opcje we współczesnej praktyce są na tyle rzadkie, że można wyciągnąć jakiekolwiek poważne wnioski.
Diagnoza zespołu Fanconiego
Oprócz wykonania wywiadu i dokładnego badania, lekarz z pewnością zaleci kilka badań w celu potwierdzenia tej choroby. Zespół Fanconiego nieuchronnie prowadzi do zaburzeń pracy nerek, co oznacza, że rutynowe badanie moczu będzie obowiązkowe. Oczywiście to nie wystarczy, aby ujawnić wszystkie cechy przebiegu choroby. Należy przyjrzeć się nie tylko zawartości białka i leukocytów w moczu, ale także spróbować wykryć lizozym, immunoglobuliny i inne substancje. Analiza koniecznie wykaże również wysoką zawartość cukru (glukozuria), fosforanów (fosfaturia), widoczne będą znaczne ubytki ważnych dla organizmu substancji. Takie badanie można przeprowadzić zarówno w warunkach ambulatoryjnych, jak i szpitalnych.

W badaniach krwi pewne zmiany są również nieuniknione. W badaniu biochemicznym odnotowano spadek prawie wszystkich znaczących pierwiastków śladowych (głównie wapnia i fosforu). Rozwija się wyraźne zakłócenie normalna operacja cały organizm.
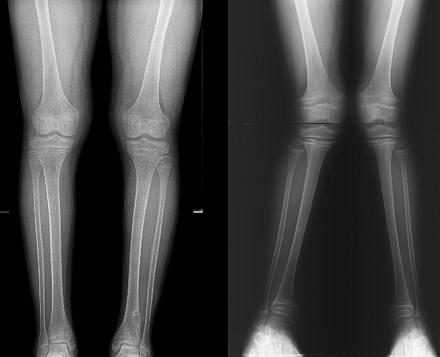
RTG szkieletu wykaże osteoporozę (zniszczenie tkanki kostnej) i deformację kończyn. W większości przypadków stwierdza się opóźnienie tempa wzrostu kości i ich niezgodność z wiekiem biologicznym. W razie potrzeby lekarz może przepisać USG nerek i inne narządy wewnętrzne, a także badanie przez powiązanych specjalistów.
Diagnoza różnicowa
Zdarzają się przypadki, gdy niektóre inne choroby podszywają się pod zespół Fanconiego. Stoi przed lekarzem trudne zadanie dowiedz się, co tak naprawdę dzieje się z małym pacjentem. Czasami mylona jest cukrzyca glukoaminofosforanowa przewlekłe odmiedniczkowe zapalenie nerek i inne choroby nerek. Zmiany w analizie moczu i cechy charakterystyczne zmiany kostne pomogą pediatrze postawić prawidłową diagnozę.
Leczenie zespołu Fanconiego
Warto wziąć pod uwagę fakt, że ta patologia jest przewlekły. Trudno się go pozbyć nieprzyjemne objawy, możesz tylko na chwilę zmniejszyć objawy choroby. Co to oferuje nowoczesna medycyna pomóc chorym dzieciom?
Dieta jest najważniejsza. Pacjentom zaleca się ograniczenie spożycia soli, a także wszystkich ostrych i produkty wędzone. Do diety dodaje się mleko i różne słodkie soki owocowe. Nie zapomnij o (śliwki, suszone morele i rodzynki). W przypadku, gdy niedobór pierwiastków śladowych osiągnął stan krytyczny, lekarze przepisują przyjmowanie specjalnych kompleksów witaminowych.
Na tle diety podaje się duże dawki witaminy D. Stan pacjenta jest stale monitorowany - co jakiś czas musi oddać krew i mocz do badań. Jest to konieczne, aby w porę wykryć rozpoczynającą się hiperwitaminozę i zmniejszyć dawkę witaminy D. Leczenie jest długie, wysokie stawki, z przerwami. W większości przypadków taka terapia pomaga przywrócić zaburzony metabolizm i zapobiec poważnym powikłaniom.

Jeśli choroba zaszła daleko, pacjent wpada w ręce chirurgów. Doświadczeni ortopedzi będą w stanie skorygować deformacje kości i znacznie poprawić standard życia dziecka. Takie operacje są wykonywane tylko w przypadku stabilnej i długotrwałej remisji: co najmniej półtora roku.
Prognoza
Niestety rokowanie dla tych pacjentów jest złe. W większości przypadków choroba postępuje powoli, prowadząc ostatecznie do niewydolności nerek. Deformacje kości szkieletowych nieuchronnie prowadzą do niepełnosprawności i pogorszenia ogólnej jakości życia.
Czy tej patologii można uniknąć? Niewątpliwie podobne pytanie niepokoi wszystkich, którzy mają do czynienia z zespołem Fanconiego. Rodzice próbują zrozumieć, co zrobili źle i gdzie nie poszli za dzieckiem. Równie ważne jest, aby wiedzieć, czy sytuacja z innymi dziećmi grozi powtórzeniem. Niestety środki zapobiegawcze nie zostały na razie opracowane. Pary planujące mieć kolejne dziecko powinny skonsultować się z genetykiem, aby uzyskać więcej informacji na temat swoich obaw.
Zespół Wisslera-Fanconiego (podsepsa alergiczna)
Ta choroba jest opisana tylko u dzieci w wieku od 4 do 12 lat. Przyczyna tej poważnej patologii jest wciąż nieznana. Można założyć, że ten syndrom jest typowy choroby autoimmunologiczne, forma specjalna reumatyzm. Zawsze zaczyna się gwałtownie, wraz ze wzrostem temperatury, która może utrzymywać się na poziomie 39 stopni przez tygodnie. We wszystkich przypadkach pojawia się polimorficzna wysypka na kończynach, czasem na twarzy, klatce piersiowej lub brzuchu. Zwykle powrót do zdrowia następuje bez poważnych komplikacji. Jednak u niektórych młodych pacjentów z czasem rozwija się poważne uszkodzenie stawów, prowadzące do niepełnosprawności.
I
Zespół De Toni - Debre - Fanconi (G. de Toni, włoski pediatra, ur. 1895; A.R. Debrt, francuski pediatra, ur. 1882; G. Fanconi, szwajcarski pediatra, ur. 1892)
Dziedziczna choroba, który opiera się na enzymatycznym niedoborze kanalików proksymalnych nerek; objawiające się głównie krzywiopodobnymi zmianami w szkielecie - patrz Choroby krzywicopodobne .
choroba dziedziczna spowodowana naruszeniem reabsorpcji fosforu, aminokwasów, węglowodanów i wodorowęglanów w kanalikach proksymalnych nerek, objawia się pod koniec pierwszego - początku drugiego roku życia z wielomoczem, niedociśnieniem, niedociśnienie tętnicze, hiporefleksja, zmiany przypominające krzywicę układ szkieletowy i spontaniczne złamania kości; dziedziczone w sposób autosomalny recesywny.
- - Olivier 1920, Paryż - 1999, Paryż. Francuski malarz i grafik. Należał do artystycznej rodziny, która rozbudziła jego wczesną pasję do malarstwa...
Sztuka europejska: malarstwo. Rzeźba. Grafika: Encyklopedia
- - Przewlekła idiopatyczna hiperkalcemia z osteosklerozą...
Słownik terminy psychiatryczne
- - miód. Zespół Fanconiego to wrodzona lub nabyta rozlana dysfunkcja proksymalnych krętych kanalików nerkowych w połączeniu z uogólnioną hiperaminoacydurią, glukozurią, hiperfosfaturią, a także ...
Podręcznik chorób
- - Ja ́ Michel, francuski mąż stanu i polityk. Prawnik. W czasie II wojny światowej brał udział w Ruchu Oporu...
- - Debre Michel, francuski mąż stanu i polityk. Prawnik. W czasie II wojny światowej brał udział w Ruchu Oporu...
Wielka radziecka encyklopedia
- - Michel - Francuz. państwo i polityczny postać. Z wykształcenia prawnik. W 1938 był pracownikiem urzędu górniczego. Finanse P. Reynaud. Na początku II wojny światowej został zmobilizowany do wojska i wkrótce został schwytany przez Niemców...
radziecki encyklopedia historyczna
- - patrz cystynoza...
Duża słownik medyczny
- - choroba dziedziczna spowodowana naruszeniem reabsorpcji fosforu, aminokwasów, węglowodanów i wodorowęglanów w kanalikach proksymalnych nerek ...
Duży słownik medyczny
- - zespół de Toni-Fanconiego - patrz zespół De Toni-Fanconiego...
Duży słownik medyczny
- - patrz Zespół Adrenogenital z utratą soli ...
Duży słownik medyczny
- - patrz cystynoza...
Duży słownik medyczny
- - patrz anemia Fanconiego...
Duży słownik medyczny
- - patrz zespół De Toni-Debre-Fanconiego...
Duży słownik medyczny
- - dziedziczne zaburzenie gospodarki wapniowo-fosforowej, objawiające się m.in dzieciństwo pozostawanie w tyle pod względem fizycznym i rozwój mentalny, liczne deformacje twarzy, zez zbieżny, zaburzenia...
Duży słownik medyczny
- - choroba, która atakuje proksymalne kanaliki kręte nefronów; może być dziedziczna lub nabyta i występuje częściej u dzieci ...
terminy medyczne
"De Toni - Debre - Syndrom Fanconiego" w książkach
Debreux Gerard (1921-2004) amerykański ekonomista urodzony we Francji
Z książki Wielkie odkrycia i ludzie autor Martyanova Ludmiła MichajłownaDebreux Gérard (1921-2004) Urodzony we Francji amerykański ekonomista Gerard Debreux urodził się w Calais jako syn Camille i Fernandy (z domu Descharne) Debreux. Zarówno jego dziadkowie, jak i ojciec byli właścicielami małej firmy koronkarskiej znajdującej się w tych okolicach
Fanconi
Z książki Coś dla Odessy autor Wasserman Anatolij AleksandrowiczFanconi Od „Szkarłatnych Żagli” przechodzimy wzdłuż Jekaterynińskiej do rogu Lanzheronowskiej. Po przeciwnej stronie ulicy Jekaterynińskiej wyraźnie widać instytucję znaną z odeskiej piosenki: Wtedy podszedł do nich słynny marker Monya, O którego kręgosłupie została złamana wskazówka w kawiarni Fanconiego.
2. Teoria równowagi ogólnej w XX wieku: wkład A. Walda, J. von Neumanna, J. Hicksa, C. Arrowa i J. Debre
Z książki History of Economic Doctrines: Lecture Notes autor Eliseeva Elena Leonidovna2. Teoria równowagi ogólnej w XX wieku: wkład A. Walda, J. von Neumanna, J. Hicksa, K. Arrowa i J. Debre Teoria równowagi ogólnej w XX wieku. rozwinął się w dwóch kierunkach, z których pierwszy może być przypisany mikroekonomii. Naukowcy związani z tym obszarem są
19.39. grandiloquence astrid lindgren i regis debré
Z księgi Stratagems. O chińskiej sztuce życia i przetrwania. TT. 12 autor von Senger Harro19.39. Natchnienie Astrid Lindgren i Régis Debre W artykule z okazji dziewięćdziesiątych urodzin światowej sławy szwedzkiej pisarki dziecięcej Gerdy Wurzenberger, oddała hołd jej zdolności zwracania uwagi opinii publicznej na poruszane przez nią kwestie niedziecięce.
Debre Michelle
TSBDebre Robert
Z książki Wielka sowiecka encyklopedia (DE) autora TSBDEBRE, Michelle
Z książki Wielki słownik cytatów i popularne wyrażenia autorDEBRE, Michel (Debr?, Michel, 1912-1996), polityk francuski 65 ... Europa jutra, Europa ojczyzn i wolności. Przemówienie na inauguracji premiera 15 stycznia. 1959? up.univ-mrs.fr/veronis/Premiers/transscript.php?n=Debre&p=1959-01-15 Wyrażenie „Europa ojczyzn” („l'Europe des patries”) wkrótce stało się
DEBRE, Michelle
Z książki Historia świata w powiedzeniach i cytatach autor Duszenko Konstantin WasiliewiczDEBRE, Michel (Debr?, Michel, 1912-1996), polityk francuski28 ... Europa jutra, Europa ojczyzn i wolności Przemówienie na inauguracji premiera 15 stycznia. 1959? up.univ-mrs.fr/veronis/Premiers/transscript.php?n=Debre&p=1959-01-15
Michel Debré (Debre, Michel, 1912-1996), polityk francuski
Z książki Słownik współczesnych cytatów autor Duszenko Konstantin WasiliewiczDEBRE Michel (Debr?, Michel, 1912-1996), polityk francuski 17 Europa ojczyzn. // Europe des patries.Z przemówienia na inauguracji premiera Francji 15 stycznia. 1959 Często mylnie przypisywany Ch
7. Pierwotna dyskineza rzęsek (zespół utrwalonych rzęsek) i zespół Kartagenera
Z książki Pediatria szpitalna: Notatki do wykładu autor Pavlova N V7. Pierwotna dyskineza rzęsek (zespół nieruchomych rzęsek) i zespół Kartagenera Na podstawie genetycznie uwarunkowanego wady strukturalnej nabłonek rzęskowy błona śluzowa dróg oddechowych Morfologiczny charakter wady w jej klasycznej wersji
46. Zespół Klinefeltera. Zespół Shereshevsky'ego-Turnera. Spermatokele. Obrzęk błon jąder i powrózka nasiennego
Z książki Urologia autor Osipova O V46. Zespół Klinefeltera. Zespół Shereshevsky'ego-Turnera. Spermatokele. opuchlizna jąder i przewód nasienny Zespół Klinefeltera to rodzaj hipogonadyzmu charakteryzujący się wrodzoną degeneracją nabłonka kanalikowego jąder o zachowanej strukturze
Zespół preekscytacji komorowej (zespół Wolfa-Parkinsona-White'a (WPW)
Z książki Dziecko Serce autor Parijska Tamara WładimirownaZespół przedwczesne pobudzenie komory (zespół Wolfa-Parkinsona-White'a (WPW) O tym zespole już wspominaliśmy, w rozdziale poświęconym napadowy tachykardia. Zajmijmy się teraz tym bardziej szczegółowo. Syndrom WPW (WPW) to stan przedwczesnego pobudzenia. To
Ann Anselin Schutzenberger Zespół przodków. Więzy międzypokoleniowe, tajemnice rodzinne, syndrom rocznicowy, przenoszenie traumy i praktyczne zastosowanie genosocjogramu
autor Schutzenberger Ann AnselinAnn Anselin Schutzenberger Zespół przodków. Więzy międzypokoleniowe, tajemnice rodzinne, syndrom rocznicowy, przekazywanie traumy i praktyczne zastosowanie genosocjogramu (przetłumaczone z francuskiego przez I.K. Masalkowa) M: Instytut Psychoterapii, 20011 (s.13)
Z książki Moje badania nad genosocjogramami i syndromem rocznicowym autor Schutzenberger Ann AnselinZ książki Ancestral Syndrome: Transgenerational Connections, Family Secrets, Anniversary Syndrome, Trauma Transfer, and the Practical Use of the Genosociogram / Per. I.K. Masalkov - Moskwa: Wydawnictwo Instytutu Psychoterapii: 2001 Do terapeutów ze szkoły w Filadelfii, która przyczyniła się
ZESPÓŁ DEJA VU ZESPÓŁ DEJA VU Od stabilności - do rozpadu czy do rozwoju? Michaił Delagin 09.05.2012
Z książki Gazeta jutro 979 (36 2012) autor Gazeta JutroNależy go odróżnić od prawdziwego zespołu w hiperwitaminozie D, w którym w wyniku enzymatycznego niedoboru aparatu kanalikowego nerek w organizmie dziecka rozwijają się ciężkie zaburzenia metaboliczne. V. V. Shitskova i in. (1971) na podstawie własnych obserwacji podają następującą różnicową tabelę diagnostyczną tych chorób (Tabela 7) z kilkoma naszymi uzupełnieniami.
| Wskaźniki | Hiperwitaminoza D | Zespół Tony'ego-Debre-Fanconiego |
| Częstotliwość | Stosunkowo często | Rzadko |
| Patogeneza | Naruszenie procesy metaboliczne, głównie wapń, z powodu przedawkowania witaminy D | Enzymopatia. Wrodzona tubulopatia. Naruszenie reabsorpcji fosforu, glukozy i azotu aminowego |
| Obraz kliniczny | Suchość i bladość skóry, pragnienie, wymioty, zaparcia, niedożywienie, nadciśnienie, powiększenie wątroby | Suchość i bladość skóry, anoreksja, pragnienie, wymioty, zaparcia, wielomocz, niedożywienie, powiększenie wątroby. Nie ma nadciśnienia. Niedociśnienie mięśniowe |
| Biochemiczne badania krwi | Hiperkalcemia w ostry okres. Fosfor jest zredukowany. Cukier i białko są normalne. Fosfataza alkaliczna nie ulega zmianie | Wapń jest normalny lub niski. Fosfor jest znacznie zmniejszony. Zmniejsza się cukier i białko, gwałtownie wzrasta aktywność fosfatazy alkalicznej. kwasica metaboliczna |
| Mocz | Reakcja Sulkowicza jest pozytywna. Proteinuria, mikrohematuria, leukocyturia. Azot aminowo cukrowy jest częściej normalny | Reakcja Sulkowicza jest negatywna. Proteinuria, fosfaturia, glukozuria, aminoacyduria |
| Radiografia kości rurkowe | Ekspansja i zagęszczanie stref wstępnego zwapnienia | Osteoporoza kości rurkowatych, strefy zwapnienia są słabe |
Zespół Fanconiego- cukrzyca aminowa. Zaburzenie metabolizmu cystyny, któremu towarzyszy glikozuria, aminoacydouri, fosfaturia, zwiększone wydzielanie alanina, utrata zasad. Jest to spowodowane dziedziczną wadą kanalików nerkowych, która uniemożliwia ponowne wchłanianie glukozy, aminokwasów i fosfatonu. Zaburzona zostaje wymiana cystyny, która odkłada się w postaci kryształów w układzie siateczkowo-śródbłonkowym, rogówce, kanalikach nerkowych i innych tkankach. Z czasem prowadzi to do postępującej niewydolności. różne funkcje kanaliki nerkowe.
Konieczne jest odróżnienie zespołu Fanconiego od cystinurii, w której cystyna nie odkłada się w tkankach. Z zespołem zdolność funkcjonalna aparat kłębuszkowy pozostaje w normie. Stężenie wapnia w surowicy jest normalne, gwałtownie spada poziom nieorganicznego fosforu. Na zdjęciu radiologicznym odnotowuje się osteomalację i rozproszone zubożenie kości alkaliami.
Zespół Fanconiego (lub de Toni-Debre-Fanconi) to choroba nerek charakteryzująca się reabsorpcją białek i jonów podczas pierwotnego stężenia moczu i związanymi z tym zaburzeniami metabolicznymi i trwałością. środowisko wewnętrzne. Ubytki w kanalikach nerkowych prowadzą do wydalania z organizmu glukozy, fosforanów, aminokwasów i wodorowęglanów.
Epidemiologia i przyczyny
Zespół Fanconiego występuje we wszystkich częściach świata, a częstotliwość jego występowania wynosi 1 na 350 000 urodzeń. Jest to dość powszechne, jeśli weźmiemy pod uwagę skalę planetarną.
Ta choroba może być wrodzona lub nabyta w wyniku progresji innych chorób. Naukowcy nie ustalili jeszcze w pełni, jaki rodzaj defektu genetycznego powoduje pojawienie się takich zmian biochemicznych. Istnieje teoria, że z powodu uszkodzenia białek tworzących błony kanalików nerkowych ich przepuszczalność jest osłabiona. Inna hipoteza mówi, że mutacja wpływa na enzymy, które ponownie wchłaniają glukozę i mikroelementy.
Przydziel pełne i niekompletny zespół Fanconiego. Jeśli wszystkie trzy składniki biochemiczne są uszkodzone, to mówią o kompletnym zespole, jeśli tylko o dwóch z trzech, to jest to zespół niepełny lub częściowy.
Czynniki ryzyka
Zespół Fanconiego rzadko występuje sam. Najczęściej wiąże się z chorobami takimi jak cystynoza, galaktozemia, glikogenoza układowa lub wrodzona, tyrozynemia i inne patologie akumulacyjne. Ponadto zatrucie fosforanami, aminoglikozydami, duże dawki tetracykliny i metale ciężkie.
Amyloidoza, niedobór witamin może być również zwiastunem zespołu. Niektórzy autorzy są skłonni sądzić, że chorobę można przypisać poważnemu stopniowi patologii przypominających krzywicę.
Patogeneza

Zespół Fanconiego lub cukrzyca glukoaminofosforanowa, która występuje częściej u autorów krajowych, rozwija się z powodu naruszenia transportu substancji przez błony kanalików nerkowych. Przyczyna tego stanu nie została jeszcze w pełni wyjaśniona przez patofizjologów, ale jej konsekwencje są dobrze znane.
Wtórne zmiany w organizmie, przypominające krzywicę, są wynikiem przedłużającej się kwasicy i obniżenia poziomu fosforu we krwi, a także zmniejszenia ilości ATP (adenozynotrójfosforanu).
Istnieje wrodzony i nabyty zespół Fanconiego. Pierwszy z reguły łączy się z fermentopatią, nietolerancją fruktozy, patologiczną akumulacją glikogenu. Zespół nabyty jest wynikiem przyjmowania toksycznych leków, takich jak chemioterapia, cytostatyki czy leki przeciwretrowirusowe. Ponadto ten zespół objawów może pojawić się po przeszczepie nerki, z amyloidozą i szpiczakiem mnogim.
Zespół Fanconiego u dorosłych

Z reguły pierwotne lub wrodzone jest wykrywane w dzieciństwie. U dorosłych zespół Fanconiego pojawia się jako choroba wtórna po poważnym zatruciu, trudnym do tolerowania leczenia. Może być również powikłaniem chorób ogólnoustrojowych.
W zespole Fanconiego glukoza, fosfor, białka i inne niezbędne substancje są wydalane z moczem pacjenta. W takim przypadku ilość utraconego płynu jest większa niż normalnie, zmniejsza się gęstość moczu. Pacjenci skarżą się na osłabienie i bóle mięśni i kości, letarg, senność.
Najczęściej wtórny zespół Fanconiego dotyka kobiety po pięćdziesięciu pięciu latach. Na tle już obniżonego poziomu wapnia dochodzi do utraty fosforu i innych pierwiastków śladowych. To wywołuje rozwój osteoporozy, aw rezultacie złamań kompresyjnych kręgów, złożonych złamań kości rurkowych. Ostatecznie pacjent może pozostać niepełnosprawny.
Objawy u dzieci
Najczęściej i wyraźnie widać zespół Fanconiego u dzieci. Objawy pojawiają się już w pierwszym roku życia i początkowo są podobne do tych u dorosłych:
- wielomocz (zwiększona ilość moczu);
- polidypsja (dziecko pije dużo płynu);
- temperatura podgorączkowa, wymioty i zaparcia.
Ze względu na ciągłą utratę witamin, pierwiastków śladowych i cukrów dziecko zaczyna opóźniać się psychicznie i rozwój fizyczny od swoich rówieśników. Lekarz może zaobserwować skrzywienie kości nóg, spadek napięcia mięśniowego, a następnie ich atrofię, do tego stopnia, że pięcioletnie dzieci nie mogą samodzielnie chodzić. Choroba postępuje z wiekiem i dojrzewanie dziecko rozwija przewlekłą niewydolność nerek.
Czasami pojawiają się również objawy choroby z innych układów, na przykład uszkodzenie oczu, centralne system nerwowy, serce i naczynia krwionośne. Można łączyć wady wrodzone układ moczowo-płciowy, narządy laryngologiczne i przewód pokarmowy.
Klasyfikacja

Klinicznie rozróżnia się odpowiednio pierwotny i wtórny zespół Fanconiego, istnieją ich typy i podgatunki.
Zespół idiopatyczny dzieli się na:
- dziedziczny;
- sporadyczne (spontaniczna mutacja u zdrowych rodziców);
- Zespół Denta.
Klasyfikacja wtórnego zespołu Fanconiego wiąże się z grupą patologii pierwotnej.
Zaburzenia metaboliczne:
- cystynoza;
- tyrozynemia, glikogenoza, galaktozemia;
- nietolerancja fruktozy.
Nabyte patologie nerek:
- paraproteinemia;
- cewkowo-śródmiąższowe zapalenie nerek;
- zespół nerwicowy;
- uszkodzenie nerek po przeszczepie;
- zespół paranowotworowy.
Zespół obserwuje się również przy zatruciu metalami ciężkimi, zatruciu truciznami organicznymi, antybiotykami, lekami toksycznymi, oparzeniami trzeciego lub czwartego stopnia.
Jak widać z powyższej klasyfikacji, utrata niezbędnych pierwiastków śladowych i białek w moczu jest rzadka niezależna choroba, z reguły jest to powikłanie innych patologii.
Diagnostyka

Objawy de Toni-Debre-Fanconiego u dzieci i dorosłych są powodem dalszych poszukiwań diagnostycznych. W analiza biochemiczna lekarz krwi może wykryć spadek wapnia, fosforu, sodu i innych pierwiastków śladowych na tle wzrostu poziomu fosfatazy alkalicznej (fosfatazy alkalicznej). Ponieważ wodorowęglany są tracone z moczem, obserwuje się utlenianie środowiska wewnętrznego. W ogólnej analizie moczu odnotowuje się obecność glukozy, fosforu, białek i aminokwasów. Mocz ma niska gęstość, to dużo.
Z metody instrumentalne Badanie rentgenowskie kości jest obowiązkowe do rozpoznania osteoporozy i deformacje patologiczne, a także badania ognisk kostnienia, w celu odróżnienia wieku kostnego od paszportowego.
Ponadto u niektórych pacjentów zaleca się wykonanie badania radioizotopowego lub scyntygrafii tkanki kostnej. Obszary aktywnego wzrostu kości będą ciemniejsze na ekranie. Umożliwi to lekarzowi określenie ciężkości choroby.
Aby określić stopień uszkodzenia tkanek nerek, wykonuje się ich biopsję. Ujawnia charakterystyczny kształt kanaliki nerek w postaci łabędziej szyi, zanik nabłonka kanalikowego, obecność obszarów zwłóknienia tkanek.
Leczenie

Jak powinien postępować lekarz po rozpoznaniu zespołu Fanconiego? Leczenie dzieci zaczyna się im szybciej, tym lepiej. Ma na celu wyrównanie nierównowagi elektrolitowej, uzupełnienie ilości białek i zasad buforowych. Pacjenci są polecani obfity napój, specjalna dieta Z wysoka zawartość mikroelementy, ograniczenie soli i wprowadzenie do diety produktów alkalizujących, takich jak mleko i soki owocowe. Ponadto dzieciom zaleca się spożywanie większej ilości suszonych owoców.
W przypadku wtórnego zespołu Fanconiego objawy niwelują się po leczeniu choroby podstawowej. Czasami, przy ciężkich deformacjach kości, może być wymagana konsultacja z chirurgami urazowymi lub ortopedami. Ale jest to możliwe tylko wtedy, gdy następuje remisja z zanikiem wszystkich objawów przez ponad półtora roku.
Profilaktyka i rokowanie
Do zapobiegania chorobie pierwotnej przed ciążą pary z zagrożoną historią rodzinną, należy poddać się poradnictwu medycznemu z zakresu genetyki. Prawdopodobieństwo pojawienia się chorego dziecka w rodzinie z tendencją do tej patologii wynosi 25 proc.
Syndrom de Tony-Debre-Fanconiego u dzieci i dorosłych kończy się rozczarowująco. występują w miąższu nerkowym nieodwracalne zmiany które prowadzą do rozwoju przewlekłej niewydolności nerek i konieczności dializy.
Zespół Fanconiego w Basenji: objawy

Niestety syndrom ten występuje nie tylko u ludzi. Zespół Fanconiego może również objawiać się u niektórych ras psów. Ponieważ wycofanie psy rasowe związane z akumulacją mutacje genetyczne, mają różne choroby dziedziczne. Jednym z nich może być zespół Fanconiego. W tym przypadku zwierzę stopniowo traci na wadze, obserwuje się zanik mięśni. Z powodu bólu kości pies staje się ospały, osłabiony, przestaje biegać i bawić się, stara się mniej nadepnąć na łapy z powodu bólu kości. Kolejnym objawem jest nadmierne pragnienie i częste oddawanie moczu.
Zespół Fanconiego (cukrzyca glukozo-fosforanowo-aminowa, choroba de Toni-Debre-Fanconiego, pierwotna izolowany zespół Fanconiego) - Choroba genetyczna, który powstał w wyniku autosomalnej mutacji recesywnej, charakteryzującej się upośledzoną reabsorpcją wody i substancji bioaktywnych z moczu pierwotnego (reabsorpcja kanalikowa), z powodu uszkodzenia kanalików nerkowych. Odnosi się do grupy chorób podobnych do krzywicy, w których występują ogólnoustrojowe zmiany metaboliczne.
Powody
Zmiany patologiczne stanowią jedną z form nadczynności przytarczyc – zaburzenia endokrynologicznego, które rozwija się wraz z nadmierną produkcją parathormonu (wytwarzanego przytarczyce) w wyniku przerostu gruczołów lub zmian nowotworowych.
Opcje dziedziczenia zespołu de Toni-Debre-Fanconiego:
- Autosomalna dominująca – wadliwy gen jest dziedziczony od jednego z rodziców (forma rodzinna).
- Autosomalny recesywny – wadliwy gen występuje u obojga rodziców. W przypadku zespołu mówimy o miejscowej formie dziedziczenia autosomalnego recesywnego (chromosom 15q15.3.)
Zespół Fanconiego u dzieci może być składnikiem innych chorób genetycznych:
- Cystynoza to nadmierne nagromadzenie cystyny (aminokwasu) w cytoplazmie (wewnętrznym płynnym środowisku komórkowym) komórki.
- - naruszenie przemiany galaktozy (monosacharydu) w glukozę, spowodowane mutacją genu odpowiedzialnego za produkcję urydylotransferazy galaktozo-1-fosforanowej (enzymu).
- Tyrozynemia typu I to niedobór hydrolazy fumaryloacetooctanu, skutkujący upośledzeniem metabolizmu tyrozyny.
- - ciężka dystrofia wątrobowo-mózgowa spowodowana zaburzeniami metabolizmu miedzi.
- fruktoza – utrata enzymu w wyniku złego wchłaniania fruktozy, jej nietolerancja, spowodowana niedoborem białka transportującego fruktozę.
Ustalono, że patologia opiera się na połączonej tubulopatii - grupie chorób, w których upośledzony jest transport substancji biologicznie czynnych w układzie kanalikowym. Głównym ogniwem w mechanizmie rozwoju zespołu Fanconiego jest defekt mitochondriów (magazyn energii komórki) w cyklu kwasów trójkarboksylowych (cykl Krebsa), który jest kluczowym etapem oddychania komórkowego.
Etapy mechanizmu rozwoju choroby, w których każdy kolejny etap będzie konsekwencją poprzedniego, można przedstawić w następujący sposób:
- Defekt mitochondrialny, tubulopatia enzymatyczna.
- Naruszenie reabsorpcji aminokwasów i enzymów w kanalikach nerkowych.
- Akumulacja kwasów ().
- Resorpcja kości (zniszczenie).
- Naruszenie odwrotnej absorpcji wapnia i potasu w kanalikach.
Komórki tracą zaopatrzenie w energię, co prowadzi do poważnych zaburzeń metabolicznych. Czynniki ryzyka obejmują:
- zatrucie metalami ciężkimi;
- toksyczne infekcje;
- przyjmowanie przeterminowanych antybiotyków tetracyklinowych;
- niedobór witaminy D;
- (naruszenie metabolizmu białek).
Klasyfikacja
Istnieją pierwotne (idiopatyczne) i wtórne formy choroby Fanconiego. Forma pierwotna rozwija się w wyniku dziedziczenia wadliwego genu. Postać wtórna występuje przy innych chorobach wrodzonych, uwarunkowanych genetycznie.
Zespół wtórny może wystąpić na tle nabytych patologii:
- paraproteinemia - obecność nieprawidłowych ciał białkowych we krwi;
- - ciężkie zaburzenia, które rozwijają się z uszkodzeniem kłębuszków nerek;
- kanalikowo-śródmiąższowe - grupa chorób nerek charakteryzujących się pierwotnym uszkodzeniem kanalików;
- nowotwór złośliwy(zespół paranowotworowy);
- w przypadku zatrucia;
- Poważne oparzenia.
Objawy
Zespół Fanconiego, którego objawy pojawiają się u dzieci do drugiego roku życia, ma dwie możliwości rozwoju, w zależności od parametrów klinicznych i laboratoryjnych:
- Pierwsza opcja jest typowa ciężki przebieg, wyraźne opóźnienie w rozwoju fizycznym, złamania i deformacje kości w wyniku hipokalcemii, złego wchłaniania w jelicie.
- Druga opcja to stosunkowo łagodny przebieg, umiarkowane oznaki opóźnienia rozwoju fizycznego, deformacje kości podczas normalny poziom wapń, wchłanianie wapnia w jelicie mieści się w normalnym zakresie.
Pierwsze objawy choroby u dzieci poniżej drugiego roku życia:
- gwałtowny spadek apetytu;
- niedobór masy ciała;
- letarg;
- (niestrawność spowodowana niedoborem energii białkowo-białkowej);
- pragnienie;
- niskie ciśnienie krwi;
- (duża liczba mocz);
- temperatura podgorączkowa;
- wymiociny.
Te dzieci nie są w stanie chodzić w wieku pięciu lat.
W szczytowym momencie choroby, w wieku pięciu lub sześciu lat, pierwszym objawem będzie (zmiękczenie kości), deformacje kości, paraliż związany z brakiem wapnia.
Po pojawieniu się pierwszych oznak następuje opóźnienie rozwoju psychicznego i fizycznego. Uogólnione (rozprzestrzeniające się po całym ciele) odwapnienie objawia się deformacjami kończyny dolne (paluch koślawy- skrzywienie wewnętrzne, szpotawość - skrzywienie zewnętrzne), utrata napięcia mięśniowego, skrzywienie skrzynia, kości przedramion i ramion. Brak fosforu u dzieci prowadzi do pojawienia się.
Wraz z postępem zespołu u dzieci, zaburzenia widzenia, choroby układu nerwowego, dróg moczowych i układ trawienny, choroby narządów laryngologicznych. Rzadko występują.
W przypadku zespołu Fanconiego u dorosłych osteomalacja występuje z powodu niedoboru minerałów i pierwiastków śladowych. Pacjenci skarżą się na ból kości, osłabienie mięśni, letarg, prawdopodobnie nasilone ciśnienie krwi, rozwój niewydolności nerek w przypadku braku terapii.
U dzieci młodym wieku nawet w pierwszych tygodniach życia mogą wystąpić objawy zespołu Wisslera-Fanconiego z powodu ciężkiego Reakcja alergiczna. Patologia charakteryzuje się gorączką, rumieniową wysypką, uszkodzeniem stawów (zwykle rąk).
Diagnostyka
Aby wykryć chorobę Fanconiego, laboratorium i metody wizualne diagnostyka. Biochemiczne badanie krwi wykazuje brak wapnia i fosforu, beta-2-mikroglobuliny (białka o niskiej masie cząsteczkowej). W moczu wykrywa się aminoacydurię (produkty metaboliczne aminokwasów), nerki (zaburzenia elektrolitowe), glikozurię (cukier w moczu), dużą ilość fosforanów, niedobór pierwiastków śladowych (sód, wapń, potas, fosfor i inne) .
odgrywa ważną rolę w ocenie czynności nerek badanie ultrasonograficzne i MRI.
Badanie rentgenowskie kości pozwala na badanie struktury kości, wykrywanie zaburzeń strukturalnych, stopnia osteoporozy, deformacji oraz ocenę opóźnienia wieku w rozwoju tkanki kostnej. W przypadku choroby Fanconiego radiografia ujawnia:
- gruba włóknista struktura kości;
- - zniszczenie nasadowej (chrzęstnej) płytki wzrostowej;
- struktura komórkowa i ostrogi narośla w kości piszczelowej;
- i złamań - w późniejszych etapach.
W badaniu emisji pozytonów (PET) wykrywa się akumulację substancji radioizotopowej w strefach wzrostu kości pacjenta.
W materiale biopsyjnym stwierdzono naruszenie struktury kości, luki (zagłębienia patologiczne), słabą mineralizację.
W cukrzycy glukozo-fosforanowo-aminowej, diagnostyka różnicowa z następującymi patologiami:
- cystynoza;
- glikogenoza;
- różne zespoły (niski, nerczycowy);
- szpiczak mnogi (złośliwa choroba krwi);
- nietolerancja fruktozy spowodowana czynnikiem genetycznym, inne choroby dziedziczne;
- stany wynikające z przeszczepu nerki.
Leczenie
Zespół Fanconiego leczy hematolog i genetyk. W przypadku nieprawidłowości w budowie struktur nerek, ciężkiego białkomoczu, konieczna jest konsultacja z urologiem i nefrologiem, z zaburzeniami endokrynologicznymi - endokrynologiem, z zaburzeniami widzenia - okulistą.
Terapia ma na celu:
- eliminacja zaburzeń elektrolitowych, w szczególności kwasicy kanalikowej;
- korekta nierównowagi kwasowo-zasadowej;
- eliminacja objawy kliniczne(terapia objawowa).
Kurs jest przepisywany preparatami potasu i wapnia, witaminą D ze stopniowym zwiększaniem dawki i dynamicznym badaniem krwi na zawartość fosforu i wapnia. Zaleca się obfite picie i dietę. W żywieniu należy ograniczyć spożycie słonych pokarmów i soli, wprowadzić do diety mleko, suszone morele, suszone śliwki, soki owocowe. Dzięki normalizacji morfologii możesz wykonywać masaże, brać kąpiele iglaste.
Interwencja chirurgiczna jest wskazana tylko w przypadku ciężkich deformacji kości. Operacja jest wykonywana ze stabilną remisją trwającą od półtora roku, co potwierdzają wskaźniki diagnostyczne i objawy kliniczne.
Bardzo poważne powikłanie Cukrzyca glukozo-fosforanowo-aminowa -. W przypadku ciężkiego stopnia choroby, która zagraża życiu pacjenta, wskazana jest hemodializa („sztuczna nerka”).

W przypadku niektórych rodzajów niewydolności nerek hemodializa jest wykonywana tymczasowo do czasu poprawy lub przywrócenia funkcji nerek. W innych przypadkach, przy nieodwracalnych procesach w nerkach, zabieg przeprowadza się na całe życie.
Dializa polega na przepuszczaniu krwi przez specjalny system, w którym z biopłynu oddzielane są substancje toksyczne, które są usuwane za pomocą roztworu dializacyjnego. Ciało zostaje uwolnione od toksycznych produktów rozpadu aż do następnej akumulacji.
Prognozy
Rokowanie zależy od choroby podstawowej, przeciwko której rozwinął się zespół. Jeśli chorobą podstawową jest nowotwór, jeśli zostanie skutecznie usunięty, rokowanie może być stosunkowo korzystne.